Newborns are a great study subject in the field of microbiology, because scientists are still discovering how the microbiome develops and what factors affect it. In human infants, it has been proven that vaginal birth exposes infants to bacteria that are different from those received by the mother through C-section. Babies born by vaginal delivery have gut bacteria correlated with vaginal bacteria, while babies born by C-section have gut bacteria correlated with human skin bacteria. For babies born by C-section, the sources of the human skin microbes that are acquired are still unknown. In a study published by Microbiome, a group of scientists tested the hypothesis that the operating room environment contains human skin bacteria that could be seeding the gut microbiome of C-section born babies.
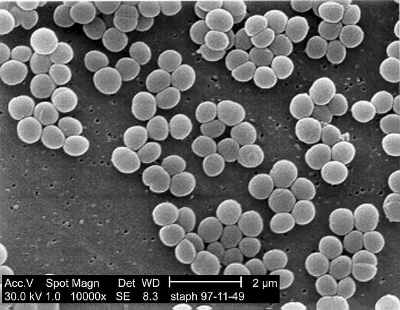
To test their hypothesis, the researchers collected samples from 11 sites in four operating rooms from three hospitals in New York City, NY and San Juan, PR. Of the 44 operating room samples that were collected, 68% of the samples contained a sufficient number of bacterial DNA samples for sequence analysis. After analyzing the bacteria collected, it was found that all samples contained human skin bacteria, with Staphylococcus and Corynebacterium being the greatest in quantity. Lamps on the operating bed and baby crib showed higher abundances of these bacteria relative to the other sampling sites. The scientists confirmed that the samples collected were more similar to human skin microbiota than other body sites, by comparing the samples to oral, fecal, and vaginal database samples.
Even though operating rooms are supposed to be spotlessly clean and germ-free, this study shows that there are still dust particles containing human skin, and therefore human skin microbiota, samples. These samples could be from people moving in and out of the operating room during a C-section, or it could come from the people cleaning the OR. Either way, the human skin bacteria in the operating room most-likely are what influences the infant’s developing gut microbiome.